In my work, I have studied desorption using two different methods: bond-boost method and temperature acceleration method. The bond-boost method is described in detail in its own section in this website. Below, I describe the temperature acceleration method with a simple example, that implicitly uses umbrella sampling of the data.
Temperature Acceleration
Desorption at low temperature is a rare event that cannot be sampled sufficiently by regular MD (over computationally tractable time period). Therefore, one way of simulating desorption is by employing accelerated MD via the temperature acceleration method. This method is based on sampling high-energy configurations using umbrella sampling. However, instead of applying a bias potential to perform high-energy sampling, the simulation is conducted at a high temperature where desorption occurs rapidly enough to allow sufficient sampling over a tractable computation period. Using the data sampled, the rate constant is calculated using the equation derived in this page:
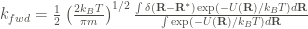
where the integral at the numerator represents the canonical average at the infinitesimally thin transition state (TS) identified by 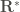

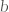
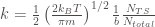
where 
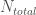
The rate constant calculated from the above setup will provide the rate constant at the high temperature employed. In order to obtain the rate constant at the original temperature, we use a weighting function 

The form of 


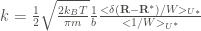
The subscript 
Let us suppose that the 
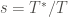

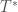
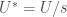
![W = \exp \left[ -\frac{s-1}{s} \frac{U(\mathbf{R})}{k_B T} \right]](https://s0.wp.com/latex.php?latex=W+%3D+%5Cexp+%5Cleft%5B+-%5Cfrac%7Bs-1%7D%7Bs%7D+%5Cfrac%7BU%28%5Cmathbf%7BR%7D%29%7D%7Bk_B+T%7D+%5Cright%5D+&bg=ffffff&fg=444340&s=0&c=20201002)
In practice, the value of 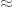
Example: 1D Desorption
Simulation
A 1D Morse potential 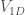
![V_{1D}(x) = D_e \left[(1-e^{-\alpha(x-x_{0})})^2-1 \right]](https://s0.wp.com/latex.php?latex=V_%7B1D%7D%28x%29+%3D+D_e+%5Cleft%5B%281-e%5E%7B-%5Calpha%28x-x_%7B0%7D%29%7D%29%5E2-1+%5Cright%5D+&bg=ffffff&fg=444340&s=0&c=20201002)

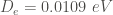
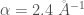
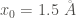
The minimum of this potential at 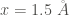



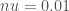
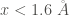


The rate constant is calculated at 25 K using the equation described in the above section.
Theoretical Calculation
In addition, the occupation probability distribution 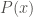
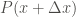

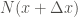


The occupation probabilities of every 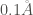

![P_{TempAcc}(x+\Delta x) = \frac{ \sum \limits_{i=1}^{N(x+\Delta x)} \exp{\left[-\frac{s-1}{s} \frac{V_{1D}(x_i)}{k_B T} \right]}}{\sum \limits_{i=1}^{N_{total}} \exp{\left[-\frac{s-1}{s} \frac{V_{1D}(x_i)}{k_B T} \right]}}](https://s0.wp.com/latex.php?latex=P_%7BTempAcc%7D%28x%2B%5CDelta+x%29+%3D+%5Cfrac%7B+%5Csum+%5Climits_%7Bi%3D1%7D%5E%7BN%28x%2B%5CDelta+x%29%7D+%5Cexp%7B%5Cleft%5B-%5Cfrac%7Bs-1%7D%7Bs%7D+%5Cfrac%7BV_%7B1D%7D%28x_i%29%7D%7Bk_B+T%7D+%5Cright%5D%7D%7D%7B%5Csum+%5Climits_%7Bi%3D1%7D%5E%7BN_%7Btotal%7D%7D+%5Cexp%7B%5Cleft%5B-%5Cfrac%7Bs-1%7D%7Bs%7D+%5Cfrac%7BV_%7B1D%7D%28x_i%29%7D%7Bk_B+T%7D+%5Cright%5D%7D%7D+&bg=ffffff&fg=444340&s=0&c=20201002)
for temperature acceleration, where 

In addition, the theoretical rate constant 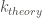
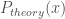
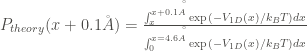
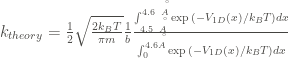

The comparison of 

Note: however, it must be pointed out that the temperature acceleration method in studying desorption is inherently flawed since the rate constant is entropy dependent, and hence temperature dependent. Therefore, by employing a large temperature, the system properties are modified and the rate constant calculated even after accounting for the bias temperature do not reflect the original rate constant. This error becomes more prominent when desorption in a system with larger degrees of freedom is studied, where entropy change from adsorption to desorption is greater, e.g. desorption of benzene (see chapter from my dissertation, pg. 31). A better method that circumvents this problem is the bond-boost method.
